Diagnosis
To diagnose biliary atresia or identify any underlying causes for persistent jaundice in infants, several tests are necessary. These include:
- Blood tests to confirm conjugated hyperbilirubinaemia
- Ultrasound scans of the liver and gallbladder to identify an atretic gallbladder
- HIDA scans (radioactive imaging of the biliary system) to confirm bile flow obstruction
- Liver biopsies to check for cirrhosis
- A surgical procedure to examine the bile ducts to assess the possibility of a bile drainage procedure for alleviating obstruction
Early diagnosis is very important. Surgery is more likely to succeed if performed before the infant reaches two months of age. The success rate diminishes significantly if the surgery occurs after the child is older than three months. Therefore, it is imperative to evaluate all infants exhibiting jaundice beyond four weeks for biliary atresia.
Treatment
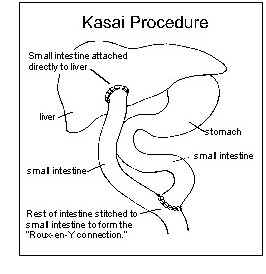
The primary treatment for biliary atresia is the Kasai Procedure. In this operation, a paediatric surgeon removes the obstructed bile ducts outside the liver and attaches a loop of the small intestine directly to the liver, allowing bile to flow into the intestine. This connection, known as a Roux-en-Y hepatoportojejunostomy, or the Kasai Procedure, can lead to the successful drainage of bile from the liver and the disappearance of jaundice. In such cases, children can live many years and grow normally.
However, if bile flow is only partially restored, the child may eventually develop cirrhosis complications and will require a liver transplant. Some children might not need a liver transplant if they experience no bile flow after the Kasai Procedure.
Adequate bile flow is needed for the digestion and absorption of dietary fats and fat-soluble vitamins such as vitamins A, B, E and K. Reduced bile flow can lead to poor growth and malnutrition in children. To counter this, special formulas containing medium-chain triglycerides (easily digested dietary fat) and water-soluble vitamin supplements are often prescribed to optimise the child's growth and development.
About 50–60% patients experience jaundice clearance after the Kasai Procedure2. Such patients can survive over 20 years with their native liver if they undergo the operation early in infancy. However, 60.5% of them may still face progressive liver-related complications1.
Long-term complications following Kasai Procedure include cholangitis and portal hypertension. Cholangitis, an infection caused by bacteria migrating up the Roux-ex-Y, presents symptoms like fever, increased jaundice and lighter stool colouration. Treatment or prevention of cholangitis requires antibiotics.
Portal hypertension, resulting from liver scarring, leads to increased pressure in the veins linking the intestines and spleen to liver. This condition can cause bleeding and clotting problems, enlarged veins in the oesophagus and stomach and ascites—an accumulation of fluid in the abdominal cavity .
When these complications become unmanageable with medication, liver transplantation comes necessary. Liver transplantation has proven highly successful in the long-term treatment of children with biliary atresia, with an overall survival of more than 90%3.
Source: 1PubMed, 2ISRN Surgery, 3PubMed
